
The European Medicines Agency (EMA) has issued a press release on 1 February 2019 with a statement that companies that make sartan blood pressure medicines, also known as angiotensin II receptor blockers, are being required to review their manufacturing processes so that they do not produce nitrosamine impurities.
Companies will have a transition period to make any necessary changes, during which strict temporary limits on levels of these impurities will apply. After this period, companies will have to demonstrate that their sartan products have no quantifiable levels of these impurities before they can be used in the EU.
These recommendations follow EMA’s review of N-nitrosodimethylamine (NDMA) and N‑nitrosodiethylamine (NDEA), which are classified as probable human carcinogens (substances that could cause cancer) and have been detected in some sartan medicines.
For the vast majority of sartan medicines, impurities were either not found or were present at very low levels.
The review estimated the highest possible cancer risk with these impurities. It concluded that if 100,000 patients took valsartan from Zhejiang Huahai (where the highest levels of impurities were found) every day for 6 years at the highest dose, there could be 22 extra cases of cancer due to NDMA over the lifetimes of those 100,000 patients. NDEA in these medicines could lead to 8 extra cases in 100,000 patients taking the medicine at the highest dose every day for 4 years. The 6 and 4 years refer to the duration of time NDMA and NDEA are believed to have been present in valsartan from Zhejiang Huahai.
The estimates have been extrapolated from animal studies and are very low compared with the lifetime risk of cancer in the EU (1 in 2).
How impurities came to be present in sartans
Before June 2018, NDMA and NDEA were not among the impurities identified in sartan medicines and were therefore not detected by routine tests.
It is now known that these impurities can form during the production of sartans that contain a specific ring structure known as a tetrazole ring under certain conditions and when certain solvents, reagents, and other raw materials are used. In addition, it is possible that impurities were present in some sartans because manufacturers had inadvertently used contaminated equipment or reagents in the manufacturing process.
Companies must now take measures to avoid the presence of these impurities and carry out rigorous testing of their products.
Testing during and after the transition period
While the goal is to have no quantifiable nitrosamine impurities in sartans, interim limits have been set for NDMA and NDEA in line with current international guidelines (International Council for Harmonisation of Technical Requirements of Pharmaceuticals for Human Use (ICH) Guidance: M7(R1)).
Products containing either impurity above these limits or products containing both nitrosamines at whatever level will not be allowed in the EU.
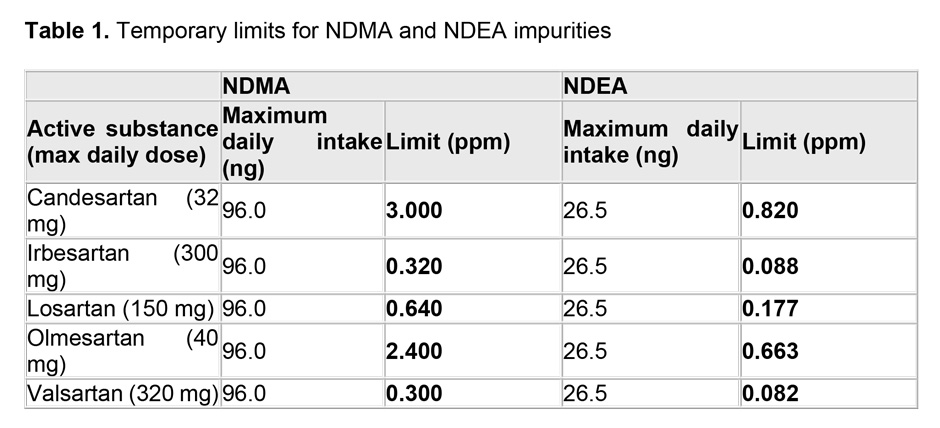
The limits are based on the maximum daily intake for each impurity derived from animal studies: 96.0 nanograms for NDMA and 26.5 nanograms for NDEA. Dividing these by the maximum daily dose for each active substance gives the limit in parts per million (see Table 1).
The transition period, which will last for 2 years, will allow companies to make the necessary changes to their manufacturing processes and to put in place testing regimes able to detect the smallest amounts of these impurities.
After the transition period, companies must exclude the presence of even lower levels of NDEA or NDMA in their products (< 0.03 parts per million).
Investigation continuing
EMA and national authorities will continue investigating the presence of nitrosamine impurities in medicines, including other impurities such as N-nitrosoethylisopropylamine (EIPNA), N-nitrosodiisopropylamine (DIPNA) and N-nitroso-N-methylamino butyric acid (NMBA).
Authorities in the EU will also consider the lessons that can be learned from this review to improve the way impurities in medicines are identified and handled.
EMA’s recommendations for NDMA and NDEA will now be sent to the European Commission for a legally binding decision. An assessment report with further details about the review will be published shortly on EMA’s website.
Information for healthcare professionals
- Nitrosamines are potent carcinogens in animals and probable carcinogens in humans.
- These impurities can form during the production of sartans that contain a tetrazole ring when certain reaction conditions are met or when contaminated materials are used.
- For NDMA, the key step involves dimethylamine (DMA) which forms the impurity in the presence of nitrites, usually under acidic conditions. A similar step – involving diethylamine (DEA) – is linked to the presence of NDEA.
- A rigorous testing regime is in place to ensure that sartan medicines are acceptably safe.
- If there is a need for further recalls or other measures, national authorities will inform healthcare professionals of what action to take.
- Manufacturers must now review their manufacturing processes to avoid the presence of nitrosamines.
More about the medicine
The review concerns candesartan, irbesartan, losartan, olmesartan and valsartan, which belong to a class of medicines called sartans (also known as angiotensin-II-receptor antagonists).
These sartan medicines have a specific ring structure (tetrazole) whose synthesis could potentially lead to the formation of nitrosamine impurities. Other medicines of the class which do not have this ring, such as azilsartan, eprosartan and telmisartan, were not included in the review.
These medicines are used to treat patients with hypertension and those with certain heart or renal diseases. They work by blocking the action of angiotensin II, a hormone that constricts blood vessels and causes blood pressure to rise.
More about the procedure
The review of valsartan medicines was triggered by the European Commission on 5 July 2018 under Article 31 of Directive 2001/83/EC. On 20 September 2018, the review was extended to include medicines containing candesartan, irbesartan, losartan and olmesartan.
The review was carried out by EMA’s Committee for Medicinal Products for Human Use (CHMP), responsible for questions concerning medicines for human use, which adopted the Agency’s opinion. The CHMP opinion will now be forwarded to the European Commission, which will issue a final legally binding decision applicable in all EU Member States.